ELA: una enfermedad neurodegenerativa sin cura (esclerosis lateral amiotrófica) ELA: una enfermedad neurodegenerativa sin cura (esclerosis lateral amiotrófica)
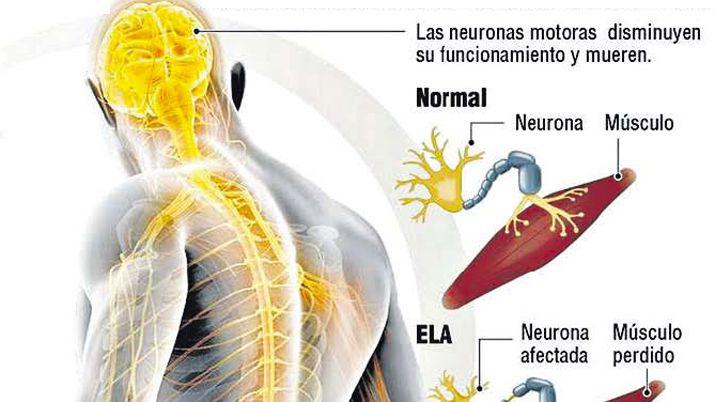
La esclerosis lateral amiotrófica (ELA) es una enfermedad degenerativa que afecta a las neuronas motoras, encargadas de controlar el movimiento de los músculos voluntarios. Afecta por tanto a los músculos de las extremidades inicialmente y hasta la parálisis, afectando con posterioridad a la voz, a la deglución y a la capacidad de respirar. Característicamente se mantiene intacta la capacidad intelectual.
De todas las enfermedades de este tipo, la ELA es la más frecuente, su incidencia aproximada es de tres de cada 100.000 personas al año, y afecta más a los varones, con una edad media de inicio de la enfermedad de unos 56 años. Actualmente es una enfermedad incurable, con una media de supervivencia en torno a los 3-5 años desde el diagnóstico. Solo un 10% de los enfermos sobreviven más de 10 años con ELA. Estos casos de mayor supervivencia suelen ser de inicio más precoz y de afectación predominantemente muscular y no bulbar. El final de la enfermedad sobreviene casi siempre por una insuficiencia respiratoria.

Existen dos formas: una familiar de origen hereditario (en el 5-10% de los casos); y una esporádica, que es la forma habitual de aparición (en el 90% de los casos), y ocurre de forma aleatoria y sin causa aparente.
Requiere de un enfoque multidisciplinar que engloba a neurólogos, neuropsicólogos, fisioterapeutas, terapeutas ocupacionales, neumólogos, nutricionistas y otros profesionales, dado que supone un alto grado de dependencia y de complicaciones.
A pesar de su carácter progresivo e irreversible, los avances científicos de los últimos años han provisto de múltiples recursos médicos, ortopédicos y sociales para minimizar las consecuencias de la enfermedad y mejorar en gran medida la vida de las personas con ELA.
CAUSAS
La causa exacta de ELA se desconoce, aunque se ha achacado a múltiples factores como el envejecimiento, alguna infección vírica, y la intoxicación por metales pesados (como el mercurio, cadmio, plomo y talio). Tan solo entre el 5% y el 10% de los casos de ELA parece deberse a causas hereditarias.
Entre los avances en ELA de los últimos años se ha enfatizado en la genética cromosómica de los casos hereditarios, atribuyendo a múltiples mutaciones (más de 100) la causa de la enfermedad.
Actualmente existen dos hipótesis, que aún no se han demostrado, pero que cabe destacar como posibles causas de ELA:
l Deficiencia de algún tipo de factor de crecimiento nervioso.
l Exceso de un neurotransmisor, denominado glutamato, en el exterior de las células del sistema nervioso.
En estos campos es donde se está haciendo hincapié en la investigación de los últimos años, para intentar identificar mejor el mecanismo íntimo que lleva a la degeneración neuronal que finalmente desemboca en la esclerosis lateral amiotrófica.
SíNTOMAS
La esclerosis lateral amiotrófica (ELA) suele comenzar a manifestarse a partir de los 50-55 años en las formas esporádicas, o bien a los 11 en las formas familiares. Se produce una debilidad muscular progresiva (que suele comenzar en una mano o un brazo), acompañada de pérdida de coordinación, que dificulta realizar actividades tan comunes como tragar, subir escaleras o levantar objetos. Entre otros síntomas, el paciente de ELA puede tener, además, calambres musculares –sobre todo tras haber realizado algún ejercicio– y algún trastorno del habla.
A medida que avanza la enfermedad, se ven involucrados más grupos musculares; hay una afectación de la musculatura distal de las extremidades (manos y pies), con fasciculaciones, exaltación de los reflejos y espasmos. Las fasciculaciones son contracciones breves e involuntarias de algún músculo, que se pueden ver por debajo de la piel y no producen ningún tipo de movimiento.
La ELA suele avanzar hacia una parálisis completa. Aunque a diferencia de otras enfermedades neurológicas, en el caso de la ELA no se presentan trastornos de la sensibilidad, afectación de esfínteres, ni pérdida de la capacidad intelectual o de la función sexual. Tampoco afecta a los músculos de los ojos, por lo que se conserva hasta el final la capacidad para realizar movimientos oculares.
En las formas bulbares las manifestaciones se centran en la mala pronunciación o disartría, que puede evolucionar a la incapacidad total para hablar. También es característica en la ELA la dificultad para tragar o disfagia, que conlleva un buen número de complicaciones como las broncoaspiraciones y la desnutrición. La afectación respiratoria también se produce en estas formas por la parálisis del diafragma condicionando una progresiva insuficiencia respiratoria que desemboca en la asistencia ventilatoria en muchos casos.
DIAGNóSTICO
Para dilucidar un diagnóstico de ELA, el neurólogo junto al neuropsicólogo elaborará una historia clínica completa, y para ello el paciente deberá responder a un interrogatorio sobre sus antecedentes quirúrgicos y enfermedades familiares, y explicar cómo se encuentra en el momento actual, aportando datos como: cuándo empezó la debilidad muscular, en qué momentos se siente peor, si existen algunos síntomas asociados como fiebre, tos, alteraciones en el ritmo intestinal, etcétera. El neurólogo realizará también un examen físico del paciente, evaluando fuerza y resistencia, y comprobando sus reflejos y la posible existencia de temblores y espasmos musculares, fasciculaciones o disminución del tejido muscular. Se observará, además, si los músculos respiratorios están afectados. Una espirometría ofrecerá información sobre el estado respiratorio del paciente.
La prueba diagnóstica que confirmará la presencia de la esclerosis lateral amiotrófica es el electromiograma, que determinará la afectación neurológica de los músculos, y demostrará la pérdida de neuronas motoras. Esta prueba consiste en colocar electrodos en un músculo y, mediante un ordenador, registrar la actividad eléctrica de cada fibra muscular.
La realización de una Resonancia Magnética Nuclear (RMN) craneal mostrará la existencia de atrofia cerebral y alteraciones en la conducción central del impulso nervioso. Esta prueba está contraindicada en pacientes con marcapasos o prótesis quirúrgicas de metal.
Deben descartarse otras enfermedades que presenten una clínica parecida; de este modo, se evitará asustar innecesariamente al paciente y a sus familiares.
A pesar de todo ello, el diagnóstico no es fácil y suele demorarse en la mayoría de casos entre 17 y 20 meses como apuntan los expertos neurólogos y neuropsicólogos.
TRATAMIENTO
La esclerosis lateral amiotrófica (ELA) es una enfermedad con un mal pronóstico, por lo que debe informarse al enfermo de su situación con tacto y detalle. En la actualidad no es posible curar la ELA, y el tratamiento va dirigido sobre todo a tratar los síntomas.
El Riluzol es un fármaco que actúa bloqueando al glutamato y se utiliza para retrasar el avance de la enfermedad y prolongar la vida. Es el único tratamiento actualmente que ha demostrado una mayor supervivencia y ha conseguido retrasar las complicaciones sobre todo de la afectación bulbar (deglución, respiración). De los múltiples fármacos que se han ensayado se ha demostrado ineficacia para antibióticos como ceftriaxona, antiinflamatorios como celecoxib, sustancias del metabolismo como la creatina, antiepilépticos como lamotrigina, gabapentina o topiramato.
Un segundo fármaco, Radicava, ha sido aprobado en el año 2017 por FDA. Esta segunda opción para el tratamiento de la ELA ha demostrado su efectividad para desacelerar los síntomas de esta enfermedad neurodegenerativa. Se trata de una infusión intravenosa que parece aportar un mayor beneficio en las primeras fases de la enfermedad, cuando acaba de ser diagnosticada y los síntomas de los pacientes son aún leves. Aunque hay que aclarar que no detiene el progreso de la ELA, y que la aprobación se ha dado tras haber sido probado en un grupo muy pequeño de pacientes. Otro inconveniente es su elevado precio, ya que seguir esta terapia ronda los 150.000 dólares anuales.
Asimismo, en el campo de la terapia génica para tratar la ELA, se investiga sobre la posibilidad de modificar el ARN con el fin de inhibir la expresión genética de SOD-1, útil solamente en los casos en los que se demostrara esta mutación. Igualmente son prometedores los estudios sobre células madre y su utilidad en este campo.
Otros medicamentos se administran a los pacientes para atenuar las molestias propias de la ELA (calambres, espasmos musculares, trastornos del sueño) y la fisioterapia y la rehabilitación se emplean para mejorar la función muscular y la movilidad de los enfermos en la medida de lo posible.
Para combatir la dificultad respiratoria y de la deglución (los músculos involucrados en estas dos funciones suelen ser los primeros en verse afectados) se pueden seguir unas pautas cuando el paciente tenga que comer:
l Permanecer con el tronco erguido y la cabeza ligeramente flexionada.
l Concentrarse en el momento de las comidas evitando, en la medida de lo posible, hablar.
l No tomar alimentos extremadamente calientes o fríos.
l Realizar comidas poco abundantes, pero con mayor asiduidad a lo largo del día.
l Masticar lentamente.
También es importante educar a los familiares del paciente sobre la enfermedad que padece. Deberán aprender a efectuar la maniobra de Heimlich para evitar posibles atragantamientos.
Esta maniobra consiste en situarse de pie por detrás de la víctima y rodearle con ambos brazos. Se sitúa la mano derecha cerrada en un puño y se coloca por debajo del esternón, la izquierda se pone abrazada al puño. Una vez preparados, se comprime rápida y fuertemente el abdomen de la víctima, desde abajo hacia arriba. Esta maniobra debe repetirse varias veces, hasta que sale con fuerza el objeto que había obstruido la vía respiratoria.
Es recomendable la intervención de un nutricionista, porque los pacientes con ELA suelen perder peso debido a las dificultades que tienen para deglutir. En algunos casos, puede ser necesario colocar una sonda en el estómago del paciente (gastrostomía) para que pueda alimentarse de acuerdo con sus necesidades sin tener que tragar los alimentos.
La musculatura respiratoria se afecta en casi todos los pacientes de ELA, pudiendo ocasionar dificultad e incluso para su respiración. Para controlar la función respiratoria del paciente deberá realizarse una espirometría cada cierto tiempo. Cuando se encuentre muy comprometida será preciso recurrir a fisioterapia respiratoria, higiene de las vías respiratorias (para evitar posibles neumonías), oxigenoterapia y, ocasionalmente, traqueotomía. En los últimos años se emplean sistemas de respiración no invasiva, como la Bipap que permite mediante una mascarilla acoplada a un respirador portátil facilitar la función ventilatoria a estos pacientes inicialmente por la noche, pero a medida que avanza la enfermedad son utilizadas también por el día.
PRONóSTICO
La esclerosis lateral amiotrófica (ELA) evoluciona hacia un empeoramiento progresivo y la media de supervivencia a partir del desarrollo de los síntomas iniciales se sitúa entre tres y cinco años, aunque alrededor del 10% de los pacientes consigue vivir más de diez años. Actualmente existen varias líneas de investigación para el desarrollo de fármacos que puedan combatir o frenar el curso de la enfermedad y mejorar el pronóstico de la ELA, como son los factores neurotróficos, antagonistas del ácido glutámico, ciclofosfamida e inmunoglobulinas intravenosas. Es posible que, en un futuro, la ELA pase a ser una enfermedad con un pronóstico más favorable. Las investigaciones en curso pretenden conseguir un tratamiento curativo o preventivo para la enfermedad, además de intentar esclarecer las causas que llevan a una persona a padecerla. l
Es una práctica cada vez más extendida, considerando que el paciente mantiene intactas sus facultades intelectuales, la posibilidad de dejar constancia del testamento vital o voluntades anticipadas (documento de instrucciones previas), al inicio de la enfermedad y una vez aportada toda la información médica disponible. De esta forma, el avance de la enfermedad será manejado médicamente de acuerdo a los deseos anticipatorios del paciente con ELA para evitar algunos tratamientos o técnicas que no se desean (traqueostomía, gastrostomía, intubación traqueal, etcétera). En este documento se nombra un representante legal del paciente que garantice el cumplimiento de dichas voluntades a lo largo de la enfermedad, siempre revisable y sujeto a modificaciones.
En la actualidad, las personas diagnosticadas de esclerosis lateral amiotrófica viven más años tras el diagnóstico de lo que lo hacían hace años, gracias a fármacos como el Riluzol, que retrasa el progreso de la enfermedad, pero también por los efectos de otros tratamientos multidisciplinares, como la rehabilitación física que mejora la capacidad muscular, la gastrostomía, que favorece una mejor nutrición, y la ventilación no invasiva, que no precisa intubación endotraqueal ni traqueotomía, por lo que permite mantener las vías respiratorias intactas, evitando riesgos como la neumonía.
Como en todas las patologías, un diagnóstico precoz y una información adecuada, que permita al enfermo y sus familiares elegir las alternativas de tratamiento y estilo de vida más favorables para su caso, mejoran la calidad de vida y las expectativas a largo plazo.







